
It has long been apparent that muscle is highly plastic and adaptive-what else is to be expected of a tissue constantly beset by a diverse range of functional demands. Far less apparent have been the underlying mechanisms. This article seeks to examine some of the factors implicated in processes of muscle growth, and, in keeping with the spirit of Mind and Muscle Magazine, elucidate the elaborate chemical pathways that transduce their effects.
But first a note on the subject matter that follows. Theory is doubtlessly the strength of science. Scientists love to devise theory, and wallow in hypothesis. They thrive on exposing gray areas, which then become the subject of new theory and experiment. Most scientists are skeptics. Paradox is evident in scientific papers, which usually proclaim outright that their topic of interest is buzzing with impenetrable contradictions that seemingly defy strict logical explanation. These maxims are firmly rooted in tradition.
However, the jungle of jargon that is their vernacular has a tendency to befuddle. Having forayed this literature, and sorted through the clutter and equivocation, I present to you, the prying reader, some perspective and insight. We'll start slow, and the pace will build quickly.
The Muscle Cell
 Skeletal muscle is one of the few tissues with the capacity for rapid and widespread repair. The source of this regenerative ability lies in precursor stem-cell reserves that are harbored by the myofibers. The myofibers (muscle fibers) that comprise skeletal muscle are basically muscle cells packed with contractile machinery (myofibrils), rechargeable energy sources (mitochondria), many nuclei (myonuclei), and a cytoplasmic unit (sarcoplasm, over two-thirds of which is water), each competing in a sense for space inside the cell [1].
Skeletal muscle is one of the few tissues with the capacity for rapid and widespread repair. The source of this regenerative ability lies in precursor stem-cell reserves that are harbored by the myofibers. The myofibers (muscle fibers) that comprise skeletal muscle are basically muscle cells packed with contractile machinery (myofibrils), rechargeable energy sources (mitochondria), many nuclei (myonuclei), and a cytoplasmic unit (sarcoplasm, over two-thirds of which is water), each competing in a sense for space inside the cell [1].
The precursor stem cells (satellite cells) are located outside the myofiber in depressions in the fiber surface between the plasma membrane (sarcolemma) and basal lamina (matrix that surrounds the muscle cells). Try to imagine a small population of little water sacs (satellite cells) sitting on top of a huge self-sealing water balloon (muscle cell), constantly dividing (mitosis), growing, becoming activated, and fusing with the balloon to spill their soggy contents and form an ever-larger one (hypertrophy of a multinucleate muscle cell).
Muscle subjected to functional overload mobilizes the mitotically-active satellite cells, in turn increasing myonuclei number of the recipient muscle cells and facilitating hypertrophy [2] -- an adaptation that will allow each nucleus to regulate more cytoplasm [3], and ultimately the enlarged muscles to undergo more forceful contractions (lift heavier loads). The functional phenotype (slow twitch fibers vs. fast and powerful muscle fibers) is actually a function of the myonuclei number of a myofiber in relation to its cross-sectional area and other variables such as ATP production methods [4], however designing a training program with adjusting this primarily in mind would be a Pyrrhic victory [5]. Check out Bodybuilding.com's workout database and create your own workout!
The compensatory adaptation is stifled, nonetheless, if satellite cells are rendered inert [6], suggesting that satellite cell mobilization is an absolutely necessary part of muscle growth. This is because myofiber nuclei are largely postmitotic (cannot divide), and the only source of nuclei is the satellite cells. Modulating the activation, proliferation, and differentiation of satellite cells are the signaling mechanisms of local and systemic growth factors [7, 8] -- primarily the local.
The concept of local and systemic response is often haunted by some confusion. Adaptation at the cellular level is very specific, since neural adaptation that accompanies activity patterns occurs at all levels of the Central Nervous System (CNS) [9, 10]. In general terms, this indicates that a resistance activity for the leg musculature does not result in adaptation in the arm musculature. Otherwise, a central mechanism would require all non-target tissues the respond accordingly. In vivo studies have instead suggested that the mechanisms that modulate cellular level adaptations reside within the impacted muscle.
GH-IGF Axis
The GH-IGF endocrine axis is a key player in mediating muscle tissue anabolism. Secreted by the acidophil cells of the anterior pituitary, circulating GH shoots to the liver where it can bind GH receptors to stimulate the expression of IGF-I and its cohort IGFBP3 (IGF binding protein 3, there are six in total), a protein that carries IGF-I and magnifies its effects. In the muscle, training may induce local increases in IFG-I independent of GH, and I will distill the importance of this shortly.
The mitogenic and myogenic effects of IGF-I are sensitive to the loading state of the muscle, ostensibly as a result of the transient elevations in serum androgen levels [11] and more significantly an upregulation in androgen receptor density [12], two important determinants of which are the intensity and volume of the resistance training bouts. The functions of IGF-I hence culminate into activating satellite cells, gene transcription and protein synthesis. As expected, activities commonly thought counterproductive to a bodybuilding lifestyle, such as endurance training, sleep deprivation, or fasting, tend to cause a decline in IGF-I levels [13].
If the GH-IGF axis is deranged, such as in patients with liver failure, a catabolic state may ensue in the form of hepatic GH resistance -- high GH, GH receptors severely downregulated, low IGF-I [14]. For those who are not familiar with this level of negative feedback, it should be pointed out that endocrine glands tend to over-secrete their hormones, but the control point for feedback inhibition is not the concentration of the hormone itself (GH) but the target (effector) in the feedback loop. So while the GH is high, it fails to have an effect on the target (IGF-I), and throws the axis out of play. Leptin resistance behaves similarly. Resistance at the receptor and postreceptor levels, together with a decreased ability to cross the blood-brain barrier, hinders the ability of serum leptin to reach its effector/target (CNS) [15].
Of course, when considering the components of any biological system, particular awareness must also be placed on the complex regulatory mechanisms ubiquitous in their inherent framework. When you block or enhance some particular aspect of seemingly linear signaling pathways, you pierce a highly interconnected network of nonlinear kinetics and feedback circuits [16]. In other words, perturb a delicately integrated human system and forget about predictable models of protein-protein interaction -- you're in for a surprise.
For instance, there is some cellular crosstalk between the IGF-I receptor and proinflammatory cytokines (immune response mediators), the former sensitive to inhibition by the latter. Because systemic inflammation plays major roles in muscle catabolism, such as the local inhibition of IGF-I signaling [17], it may seem paradoxical that proinflammatory cytokines also stimulate the proliferation of satellite cells in vitro [18]. In fact, it is in a concerted fashion that IGF-I activates the so-called JAK and STAT proteins [19, 20] to suppress cytokine signaling and keep the inflammatory response constantly in check.
Upon closer inspection, however, it can be seen that the inflammatory response actually plays a pivotal role in muscle adaptation [21], albeit a counterproductive role when one excessively transitions into overtraining [22]. Such examples, and numerous others that exhibit comparable ingenuity, should demonstrate that a logical fallacy is committed in presuming that antagonists of anabolic signals are by default catabolic by all accounts, and vice-versa. This sort of tight coupling between communication and regulation exists in many biological contexts, underlining the importance of a multitargeted, integrative approach to understanding and dominating physiological dynamics.
At any rate, IGF-I exists in multiple isoforms (tissue-specific proteins of functional and structural similarity). One isoform, which differs from the systemic or liver type, happens to be particularly sensitive to mechanical signals such as the gamut of exercise overload. This important isoform, called mechano-growth factor (MGF), is a local splice variant of IGF-I produced by damaged or loaded skeletal muscle [23].
MGF does not circulate in the blood, but instead acts on locally targeted muscle tissue as an autocrine/paracrine signal, thereby stimulating the proliferation of satellite cells and increasing protein production in muscle fibers. For purposes of clarification, autocrine and paracrine substances act on the cell that secreted them and neighboring cells, respectively, without entering the bloodstream. As such, locally produced growth factors supplement the systemically circulating IGF-I as potent growth-inducing agents. Since muscle growth is for the most part a local phenomenon, locally produced growth factors are of central importance, whereas systemically derived IGF-I is relegated to roles in carbohydrate and fat metabolism [24].
Interestingly enough, MGF gene transfer with gene therapy, such as the coupling of the MGF gene with a viral delivery vector, would be a rather safe doping technique precisely because the effects are localized, and blood/urine samples would prove futile in a screening test. Is it possible that one day the athletic edge will come in the form of an injectable gene? Ethical considerations abound [25], but it is widely speculated that genetic manipulation is already underway, and will be nearly impossible to detect.
Essentially, what we are starting to discern here is a host of intricate modes of interaction at the cellular level of the muscle -- principally, to cope with disruptions to homeostasis thus far referred to as "loading" and "exercise". As we dig deeper into this rabbit hole, try not to lose focus of the big picture -- we are agitating a gang of muscle cells with all sorts of peevish stimuli, and in response they bark at us with biochemical signals.
Biochemistry 101
 In order to make sense of the reversible phosphorylation/dephosphorylation processes in the discussions that follow, it should be understood that in a biological system, the phosphorylation state of proteins can function as an on-off switch vis-�-vis their activity [26].
In order to make sense of the reversible phosphorylation/dephosphorylation processes in the discussions that follow, it should be understood that in a biological system, the phosphorylation state of proteins can function as an on-off switch vis-�-vis their activity [26].
Phosphorylation is a type of posttranslational modification that tags a protein with a phosphate, allowing the regulated protein to switch states in response to stimuli that require rapid adjustments of their function. Although phosphorylation can serve to either activate or inactive a protein, depending on the context, this effective form of regulation is one powerful way for the body to control such processes as cell division, gene expression, metabolism, biosynthesis cascades, and so forth. For instance, if a cultured cell is repeatedly exposed to epinephrine, several residues of the beta-adrenergic receptor become phosphorylated and hence desensitized.
A feedback loop then provides an opportunity for the phosphorylated receptors to be dephosphorylated and resensitized when epinephrine is removed [27]. Enzymes that phosphorylate proteins are called kinases, whereas those that remove phosphates are called phosphatases -- so don't fret when you encounter them in disguise.
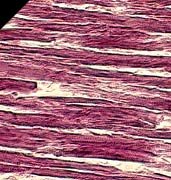
Calcineurin Pathway
Calcineurin can be considered in simple terms as a multifunctional enzymatic switch. As a calcium-sensitive phosphatase, it dephosphorylates a family of transcription factors called NFAT (nuclear factor of activated T cells). After a sustained series of intracellular calcium spikes, which calmodulin (calcium sensor and subunit of calcineurin) perceives as a prolonged calcium stimulus, dephosphorylation by the cytoplasmic calcineurin ensues to activate the NFATs by unmasking the local signals that induce their movement into the nucleus, where they can then can bind DNA sites and control gene expression [28].
As a corollary, scientists noted the opportunity for NFAT activity to be used as a gauge of calcineurin activity. By administering Cyclosporin A (CsA), a calcineurin-inhibitor, they could assess the intriguing roles of these pathways in muscle hypertrophy.
Typically, when a muscle is subject to workload increase via the surgical removal of its synergist, it will compensate within several weeks by increasing in size and strength [29]. (A synergist is a muscle that assists a prime mover, such as the front deltoids assisting the chest in the bench press). Scientists found that this compensatory hypertrophy is inhibited with CsA, and concluded that calcineurin is thus a requirement for hypertrophy [30]. Other studies, however, completely blocked calcineurin-mediated NFAT activation but did not see an inhibition of hypertrophy [31, 32].
The confusion and conflict between these studies is the result of experimental design. In the first study, the calcinueurin blockers were administered to undifferentiated myoblasts, before differentiation and fusion into multinucleated myotubes could take place. This is significant because these two stages of myogenesis respond differently to IGF-I: pre-differentiated myoblasts proliferate [33], whereas myotubes hypertrophy [34]. As I pointed out in the beginning, the hypertrophy of mammalian skeletal muscle concerns itself primarily with the fusion of differentiated muscle cells. In short, the pharmacologic inhibition of calcineurin blocks differentiation, not hypertrophy.
In fact, calcineurin activity does not increase during a load-induced hypertrophy process. It actually decreases, and hyper-phosphorylation (deactivation) of NFAT follows. In other words, IGF-I inhibits calcineurin, because calcineurin would otherwise start killing myocytes [35]. It was mentioned earlier that a family of proteins paradoxically stimulated myoblast proliferation in vitro -- proinflammatory cytokines. It was also mentioned that calcineurin-mediated NFATs regulate gene expression -- cytokine genes and other genes critical for the immune response. That CsA drug -- an immunosuppressant.
PI3K/Akt Pathways
With the calcineurin conundrum now largely pieced together, one notable question remains -- with a direct role in skeletal muscle hypertrophy per se relinquished, why is it even of concern? The answer should not come as a surprise. It turns out that the PI3K/Akt signaling pathway is a key transducer of IGF-I-mediated adaptive hypertrophy, and it functions in part by antagonizing calcineurin [36].
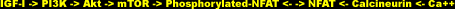
Besides IGF-I, other factors activating the PI3K/Akt pathway include cell swelling, insulin, certain amino acids, and androgens. Although interesting in their own right, our focus rests largely with the actions of IGF-I, since its local effects are of greatest concern. Numerous pathways are often simultaneously activated in response to growth factors and hormones. The binding of IGF-I to its receptor results in the phosphorylation of insulin receptor substrate 1 (IRS-1), IGF-I's major substrate. IRS-I can be thought of simply as a signaling adaptor that recruits multiple other intracellular signaling proteins [37].
One recruit that it enrolls is phosphatidylinositol 3-kinase (PI3K). One of the primary functions of PI3K activation is the resulting synthesis of a phospholipid that can activate a protein kinase called Akt [38], also called Protein Kinase B. Once it makes its way to the membrane for phosphorylation/activation through some fancy lipid-protein interactions, Akt follows in kind to activate a host of downstream targets that regulate protein synthesis and ultimately the hypertrophy of skeletal muscle cells. Mice lacking the gene to produce this protein kinase experience growth retardation and increased spontaneous cell death in the testes [39].
The immediate downstream target of Akt is the so-called Mammalian Target of Rapamycin (mTOR), a kinase forever stuck with a stodgy name and nifty acronym that probably originated as a joke of cruel simplicity. By now, if you are still with me, you might be asking yourself, "does this mTOR kinase do anything other than phosphorylate other kinases?" The answer is a resounding yes. Aside from its phosphorylation duties, mTOR is a detector of intracellular ATP levels, it is an ATP sensor. In fact, its activity is dependent on the ATP concentration, and is highest when ATP levels are correspondingly high [40].
 Far be it from me to resist a comparison. Let me briefly introduce another ATP sensor and regulator of energy metabolism that should sound rather familiar: AMPK, the ultra sensitive AMP-activated protein kinase that is activated by a drop in the homeostatic ATP/AMP ratio [41]. It has been shown, and makes logical sense, that AMPK activity downregulates mTOR activity -- an indirect effect of ATP levels falling [42]. Any stress environment that disturbs the crucial ATP balance, such as glucose deprivation, metabolic stressors, or oxygen deprivation, activates AMPK.
Far be it from me to resist a comparison. Let me briefly introduce another ATP sensor and regulator of energy metabolism that should sound rather familiar: AMPK, the ultra sensitive AMP-activated protein kinase that is activated by a drop in the homeostatic ATP/AMP ratio [41]. It has been shown, and makes logical sense, that AMPK activity downregulates mTOR activity -- an indirect effect of ATP levels falling [42]. Any stress environment that disturbs the crucial ATP balance, such as glucose deprivation, metabolic stressors, or oxygen deprivation, activates AMPK.
Exercise does all three, and the activation of AMPK is a result of ATP consumption [43]. When activated, the job of AMPK is to switch off the anabolic ATP-consuming activities in favor of catabolic ATP-producing activities. The glucose- and fatty acid- derived ATP is necessary to restore the body's sense of balance and homeostasis. Accordingly, mTOR activity is low during exercise, but rebounds with rest and feeding as AMPK fades out of play and mTOR flips its switch into the anabolic direction.
Here's how mTOR relays the signal to kick up the hypertrophy. First, it phosphorylates/activates the positive regulator p70 S6 kinase (p70S6K) sub pathway [44]. Activated/phosphorylated p70S6k phosphorylates the protein S6 in the 40S ribosomal subunit [45], an event known to correlate with the increased protein synthesis following growth factor stimulation [46]. Next, mTOR phosphorylates/deactivates the initiation factor 4E binding protein (4E-BP1) [44]. Here's why. When dephosphorylated, 4E-BP1 binds and inhibits the initiation factor eIF4E.
Because the available of eIF4E is a rate-limiting step in protein synthesis [47], this binding puts the breaks on the translational machinery. Phosphorylated 4E-BP1, however, dissociates from eIF4E and is unable to bind back, liberating the initiation factor to initiate the translation process. Scientists term this latter phenomenon negative regulation (-I), as opposed to the positive regulation (->�) seen with p70S6K. In sum, mTOR activity activates the activators and deactivates the repressors of the protein synthesizing machinery downstream of it:

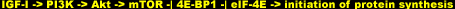
Rapamycin, and immunosuppressant and curiously aforementioned mTOR inhibitor, has been of value in experimentally elucidating this connection between mTOR signaling and hypertrophy. Treatment with rapamycin decreased muscle growth [48] and hypertrophy [34, 49] in myotube cultures in vitro. But not entirely, suggesting that other signaling pathways are involved. Here's how researchers came to such conclusions. Although we tend to hear only about mice with knockout genes, another experimental trick is to use a constitutively active form of a gene.
A constitutively expressed gene is always expressed and cannot be shut down by any of the cell's regulatory mechanisms. By comparing hypertrophic responses with constitutively expressed genes and their downstream targets, one can pinpoint the levels at which signaling pathways branch out, how the branching distributes the signal, and the propensity to which the strength of the signal is divided.
The constitutively active form of Akt causes a more potent hypertrophic response than the constitutively active form of p70S6K [49], a result consistent with the idea that p70S6K is only one of several important branched-out sub pathways downstream of Akt. The other one, as we just saw, is the 4E-BP1 sub pathway. Yet another is the glycogen synthase kinase 3 (GSK3) sub pathway, which is phosphorylated/inhibited immediately downstream of Akt in a negative regulation manner similar to that of mTOR and the initiation factor binding protein. GS3K inhibits initiation factor eIF2B; the inhibition of GS3K by Akt signaling thereby activates eIF2B and promotes protein synthesis aloof from mTOR:
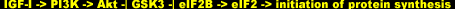
Ras/Raf/MEK/ERK Pathway & Mechanotransduction
Ras is a small GTPase switch protein that sits anchored to the cytoplasmic face of the plasma membrane and helps relay signals from the cell surface to the nucleus. Ras is often discussed in the context of cancer, since mutated forms of growth factor receptors can send growth signals unrelentingly, causing uncontrollable growth. But our focus will turn to its role in skeletal muscle, a role that is inherently similar.
In analogous fashion to that of IRS-1 described earlier, a complex of two proteins called GRB2 and Sos act as adapters to transmit a signal from IGF-I to Ras. The activation of Ras is the critical step that triggers a cascade of protein kinases that operate sequentially. First, Ras binds to Raf (mitogen-activated protein kinase kinase kinase, MAPKKK), which in turn binds to and phosphorylates MEK. MEK (mitogen-activated protein kinase kinase, MAPKK), is a dual specificity kinase that phosphoryles ERK at two residues (tyrosine and threonine).
Activated/phosphorylated ERK (mitogen-activated protein kinase, MAPK) goes on to phosphorylate a host of proteins including nuclear transcription factors. The word mitogen simply denotes any compound, such as a growth factor or hormone that induces the proliferation or growth of a tissue. In our case, the mitogen is IGF-I. The following two schematic representations are one and the same, serving only to clarify the ambiguity in the nomenclature:
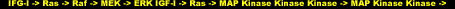
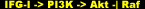
The Ras/Raf/MEK/ERK (Ras/MAPKKK/MAPKK/MAPK) pathway is distinct from the PI3K/Akt pathway, but is similarly involved in membrane-to-nuclear stress/nutrient-sensing signaling events that consummate to trigger a cascade of responses collectively leading to skeletal muscle hypertrophy. Simultaneously activated pathways often additionally undergo cross-regulation, and that will be the recurring theme for this final pathway.
When scientists, always up to their experimental tricks, began manipulating these pathways during muscle differentiation, they noticed that inhibiting Ras/Raf/MEK/ERK promotes differentiation [50], whereas inhibition of PI3K/Akt blocks it [51]. In differentiated myotubes, activated PI3K/Akt inhibited Ras/Raf/MEK/ERK [34], yet IGF-I activated both pathways [52]. Together with these results comes another experimental outcome. It has been evidenced that the Ras/Raf/MEK/ERK pathway inhibits the development of the hypertrophic phenotype of myotubes in vitro, and, through negative regulation, activated Akt is able to phosphorylate and terminate Raf kinase activity to reassert a favorable stance toward the hypertrophic response.
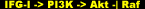
The Ras/Raf/MEK/ERK cascade just described happens to be one of mechanotrandsuction, the fundamental process by which the muscle cell perceives mechanical stress in terms of intracellular signals. There exists a relationship between peak tension and MAP Kinase phosphorylation, suggesting that the phosphorylation of MAPK can be used a quantitative marker of the mechanical stress applied to the muscle [53]. That is, the contraction/stretch components of a repetition both upregulate transcription factors, and it is signaling through the MAPK proteins that facilitates this action, which begins to show effect within minutes of growth factor stimulation.
The obvious question at present time is one that wonders why IGF-I would trigger multiple cascades of contradictory purpose. Additionally, why activate multiple pathways that inhibit each other? The answer has to do with our recurring theme of fine-tuned regulation permeating every aspect of intracellular communication -- contingencies put in place through evolution to deal with partial disruptions. The system makes no claims to perfection. Frankly, it could use a tune-up. But it does the job with remarkable efficiency, so it warrants some consideration.
Another question that seems appropriate is one that asks how exactly the Ras/Raf/MEK/ERK pathway opposes the hypertrophic response, if its end-result is also that of protein synthesis. The clinching answer ties together the thesis advanced, recapitulated, and elaborated throughout this paper. The answer, that is, has to do with the type of proteins that are synthesized as a result of MAP Kinase activation. Having reached the crescendo, such will be the following concluding section.
Endgame
Get ready for some groovy acronyms. The MAP Kinase family can be classified into two major subfamilies: ERKs (extracellular signal-regulated protein kinases) and SAPKs (stress-activated protein kinases) [54]. As their name indicates, stress activated protein kinases (SAPKs), such as the c-Jun N-terminal kinase (JNK) and the p38 MAPK [55], are kinases conserved through evolution to respond to numerous cellular stresses. Exercise being our form of stress, SAPK and ERK activity responds accordingly [56].
Eccentric contractions that lengthen the muscle increase JNK activity to a greater extent (15-fold vs. 3.5-fold) than concentric contractions [57], consistent with the idea that the greater source of mechanical stress in lengthening contractions leads to more profound adaptations in muscle morphology [58]. With stretch stimulus comes large JNK activity and p38 phosphorylation (20-fold increase above basal), but ERKs have been found to be less dramatically phosphorylated (only 2-fold increase above basal) [59].
 Curiously, researchers noted these effects in the absence of systemic factors, complementing a theory that mechanical tension alone is the bridge between exercise and the cellular adaptation response. Overlooked, however, was the importance of the autocrine/paracrine local response, with alternate pathways that concern themselves more directly with the early hypertrophic response.
Curiously, researchers noted these effects in the absence of systemic factors, complementing a theory that mechanical tension alone is the bridge between exercise and the cellular adaptation response. Overlooked, however, was the importance of the autocrine/paracrine local response, with alternate pathways that concern themselves more directly with the early hypertrophic response.
Current evidence indicates a direct role for these major MAPK pathways in mediating the effects of pro-inflammatory cytokines [60]. These kinases phosphorylate, through multi-step cascades, transcription factors belonging to families of mediators that regulate stages of cytokine synthesis and bioavailability. JNK and p38 are also on the receiving end, responding to these pro-inflammatory cytokines, such as TNF-alpha and IL-1 [61], in addition to the other forms of cellular stress. In an almost self-perpetuating manner, these SAPKs lead to the synthesis of more pro-inflammatory cytokines.
The ERK group, activated via the Ras pathway in response to growth factors [62] and exercise [56], translocates to the nucleus where it phosphorylates transcription factors [63], thereby increasing their transcription rate, and enhances the ability of eIF4E to recruit protein-synthesizing ribosomes. The ERK pathway can be stimulated and modulated by intracellular calcium levels as well. Since the calcineurin pathway also responds to a calcium stimulus, it too mediates the activation of ERK. In contrast to the SAPKs, however, the ERK cascade is in some part protective, anti-apoptotic, and conducive to cell growth, unlike the damaging stressors. Interestingly, the popular class of drugs known as NSAIDs (non-steroidal anti-inflammatory drugs), in addition to blocking prostaglandin synthesis, attenuate the proinflammatory cytokine- induced phosphorylation of ERK [64].
Remember that cellular crosstalk between IGF-I and proinflammatory cytokines? The downregulation of IGF-I by pro-inflammatory cytokines occurs through the JNK pathway [65]. As described earlier, IGF-I calls upon the JAK/STAT pathway to counter this. Multi-purpose pathways converge to ensure cross-regulation. In concert, extensive cross-talk is what enables a cell to integrate information from multiple signals and function normally in the face of disruptions to one component of a vast network. It is probable that, through evolution, proteins have learned to associate signal combinations with specific functions.
And that brings us to an exciting conclusion. Much as this may have stung the cerebral, it highlights the remarkable complexity of molecular science. An important first step in understanding any cellular process is to identify the molecular players involved.
Author's Note: You can correctly assume that any errors in this article were inserted purposely to see if you were paying attention. Do not report them. For all other comments: gene@avant-research.com
This article appears courtesy of www.mindandmuscle.net
References
[1] Lindstedt SL, McGlothlin T, Percy E, Pifer J. Task-specific design of skeletal muscle: balancing muscle structural composition. Comp Biochem Physiol B Biochem Mol Biol. 1998 May;120(1):35-40.
[2] Allen DL, Monke SR, Talmadge RJ, Roy RR, Edgerton VR. Plasticity of myonuclear number in hypertrophied and atrophied mammalian skeletal muscle fibers. J Appl Physiol. 1995 May;78(5):1969-76.
[3] Allen DL, Roy RR, Edgerton VR. Myonuclear domains in muscle adaptation and disease. Muscle Nerve. 1999 Oct;22(10):1350-60.
[4] Edgerton VR, Roy RR. Regulation of skeletal muscle fiber size, shape and function. J Biomech. 1991;24 Suppl 1:123-33.
[5] Shoepe TC, Stelzer JE, Garner DP, Widrick JJ. Functional adaptability of muscle fibers to long-term resistance exercise. Med Sci Sports Exerc. 2003 Jun;35(6):944-51.
[6] Rosenblatt JD, Yong D, Parry DJ. Satellite cell activity is required for hypertrophy of overloaded adult rat muscle. Muscle Nerve. 1994 Jun;17(6):608-13.
[7] Florini JR, Ewton DZ, Coolican SA. Growth hormone and the insulin-like growth factor system in myogenesis. Endocr Rev. 1996 Oct;17(5):481-517.
[8] Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol. 2001 Aug;91(2):534-51. Review. Erratum in: J Appl Physiol 2001 Dec;91(6):2414.
[9] Duchateau J, Enoka RM. Neural adaptations with chronic activity patterns in able-bodied humans. Am J Phys Med Rehabil. 2002 Nov;81(11 Suppl):S17-27.
[10] Carroll TJ, Riek S, Carson RG. Neural adaptations to resistance training: implications for movement control. Sports Med. 2001;31(12):829-40.
[11] Kraemer WJ. Endocrine responses to resistance exercise. Med Sci Sports Exerc. 1988 Oct;20(5 Suppl):S152-7.
[12] Bamman MM, Shipp JR, Jiang J, Gower BA, Hunter GR, Goodman A, McLafferty CL Jr, Urban RJ. Mechanical load increases muscle IGF-I and androgen receptor mRNA concentrations in humans. Am J Physiol Endocrinol Metab. 2001 Mar;280(3):E383-90.
[13] Friedl KE, Moore RJ, Hoyt RW, Marchitelli LJ, Martinez-Lopez LE, Askew EW. Endocrine markers of semistarvation in healthy lean men in a multistressor environment. J Appl Physiol. 2000 May;88(5):1820-30.
[14] Picardi A, Gentilucci UV, Zardi EM, Caccavo D, Petitti T, Manfrini S, Pozzilli P, Afeltra A. TNF-alpha and Growth Hormone Resistance in Patients with Chronic Liver Disease. J Interferon Cytokine Res. 2003 May;23(5):229-35.
[15] Banks WA, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity is acquired and reversible. Am J Physiol Endocrinol Metab. 2003 Jul;285(1):E10-5. Epub 2003 Mar 04.
[16] Bray D. Intracellular signalling as a parallel distributed process. J Theor Biol. 1990 Mar 22;143(2):215-31.
[17] Fernandez-Celemin L, Pasko N, Blomart V, Thissen JP. Inhibition of muscle insulin-like growth factor I expression by tumor necrosis factor-alpha. Am J Physiol Endocrinol Metab. 2002 Dec;283(6):E1279-90.
[18] Cantini M, Massimino ML, Rapizzi E, Rossini K, Catani C, Dalla Libera L, Carraro U. Human satellite cell proliferation in vitro is regulated by autocrine secretion of IL-6 stimulated by a soluble factor(s) released by activated monocytes. Biochem Biophys Res Commun. 1995 Nov 2;216(1):49-53.
[19] Gual P, Baron V, Lequoy V, Van Obberghen E. Interaction of Janus kinases JAK-1 and JAK-2 with the insulin receptor and the insulin-like growth factor-1 receptor. Endocrinology. 1998 Mar;139(3):884-93.
[20] Zong CS, Chan J, Levy DE, Horvath C, Sadowski HB, Wang LH. Mechanism of STAT3 activation by insulin-like growth factor I receptor. J Biol Chem. 2000 May 19;275(20):15099-105.
[21] Vierck J, O'Reilly B, Hossner K, Antonio J, Byrne K, Bucci L, Dodson M. Satellite cell regulation following myotrauma caused by resistance exercise. Cell Biol Int. 2000;24(5):263-72. Review.
[22] Smith LL. Cytokine hypothesis of overtraining: a physiological adaptation to excessive stress? Med Sci Sports Exerc. 2000 Feb;32(2):317-31.
[23] Hameed M, Orrell RW, Cobbold M, Goldspink G, Harridge SD. Expression of IGF-I splice variants in young and old human skeletal muscle after high resistance exercise. J Physiol. 2003 Feb 15;547(Pt 1):247-54.
[24] Isaksson OG, Jansson JO, Sjogren K, Ohlsson C. Metabolic functions of liver-derived (endocrine) insulin-like growth factor I. Horm Res. 2001;55 Suppl 2:18-21.
[25] Murray TH. Reflections on the ethics of genetic enhancement. Genet Med. 2002 Nov-Dec;4(6 Suppl):27S-32S.
[26] Cohen P. Protein phosphorylation and hormone action. Proc R Soc Lond B Biol Sci. 1988 Jul 22;234(1275):115-44.
[27] Hausdorff WP, Caron MG, Lefkowitz RJ. Turning off the signal: desensitization of beta-adrenergic receptor function. FASEB J. 1990 Aug;4(11):2881-9.
[28] Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707-47.
[29] Gardiner P, Michel R, Browman C, Noble E. Increased EMG of rat plantaris during locomotion following surgical removal of its synergists. Brain Res. 1986 Aug 13;380(1):114-21.
[30] Dunn SE, Burns JL, Michel RN. Calcineurin is required for skeletal muscle hypertrophy. J Biol Chem. 1999 Jul 30;274(31):21908-12.
[31] Serrano AL, Murgia M, Pallafacchina G, Calabria E, Coniglio P, Lomo T, Schiaffino S. Calcineurin controls nerve activity-dependent specification of slow skeletal muscle fibers but not muscle growth. Proc Natl Acad Sci U S A. 2001 Nov 6;98(23):13108-13.
[32] Dupont-Versteegden EE, Knox M, Gurley CM, Houle JD, Peterson CA. Maintenance of muscle mass is not dependent on the calcineurin-NFAT pathway. Am J Physiol Cell Physiol. 2002 Jun;282(6):C1387-95.
[33] Svegliati-Baroni G, Ridolfi F, Di Sario A, Casini A, Marucci L, Gaggiotti G, Orlandoni P, Macarri G, Perego L, Benedetti A, Folli F. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology. 1999 Jun;29(6):1743-51.
[34] Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999 Nov 26;286(5445):1738-41.
[35] Musaro A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999 Aug 5;400(6744):581-5.
[36] Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001 Nov;3(11):1014-9.
[37] Myers MG Jr, Sun XJ, White MF. The IRS-1 signaling system. Trends Biochem Sci. 1994 Jul;19(7):289-93.
[38] Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002 Jul;2(7):489-501. Review. No abstract available.
[39] Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001 Sep 1;15(17):2203-8.
[40] Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science. 2001 Nov 2;294(5544):1102-5.
[41] Ponticos M, Lu QL, Morgan JE, Hardie DG, Partridge TA, Carling D. Dual regulation of the AMP-activated protein kinase provides a novel mechanism for the control of creatine kinase in skeletal muscle. EMBO J. 1998 Mar 16;17(6):1688-99.
[42] Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002 Jul 5;277(27):23977-80.
[43] Winder WW. Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle. J Appl Physiol. 2001 Sep;91(3):1017-28.
[44] Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP, Kasuga M, Nishimoto I, Avruch J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem. 1997 Oct 17;272(42):26457-63.
[45] Chou MM, Blenis J. The 70 kDa S6 kinase: regulation of a kinase with multiple roles in mitogenic signalling. Curr Opin Cell Biol. 1995 Dec;7(6):806-14.
[46] Dufner A, Thomas G. Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res. 1999 Nov 25;253(1):100-9.
[47] Gautsch TA, Anthony JC, Kimball SR, Paul GL, Layman DK, Jefferson LS. Availability of eIF4E regulates skeletal muscle protein synthesis during recovery from exercise. Am J Physiol. 1998 Feb;274(2 Pt 1):C406-14.
[48] Pallafacchina G, Calabria E, Serrano AL, Kalhovde JM, Schiaffino S. A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc Natl Acad Sci U S A. 2002 Jul 9;99(14):9213-8.
[49] Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001 Nov;3(11):1009-13.
[50] Bennett AM, Tonks NK. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997 Nov 14;278(5341):1288-91.
[51] Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997 Mar 7;272(10):6653-62.
[52] Avruch J. Insulin signal transduction through protein kinase cascades. Mol Cell Biochem. 1998 May;182(1-2):31-48.
[53] Martineau LC, Gardiner PF. Insight into skeletal muscle mechanotransduction: MAPK activation is quantitatively related to tension. J Appl Physiol. 2001 Aug;91(2):693-702.
[54] Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci. 1995 Mar;20(3):117-22.
[55] Sluss HK, Barrett T, Derijard B, Davis RJ. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol Cell Biol. 1994 Dec;14(12):8376-84.
[56] Wretman C, Lionikas A, Widegren U, Lannergren J, Westerblad H, Henriksson J. Effects of concentric and eccentric contractions on phosphorylation of MAPK(erk1/2) and MAPK(p38) in isolated rat skeletal muscle. J Physiol. 2001 Aug 15;535(Pt 1):155-64.
[57] Boppart MD, Aronson D, Gibson L, Roubenoff R, Abad LW, Bean J, Goodyear LJ, Fielding RA. Eccentric exercise markedly increases c-Jun NH(2)-terminal kinase activity in human skeletal muscle. J Appl Physiol. 1999 Nov;87(5):1668-73.
[58] Goldspink G, Scutt A, Martindale J, Jaenicke T, Turay L, Gerlach GF. Stretch and force generation induce rapid hypertrophy and myosin isoform gene switching in adult skeletal muscle. Biochem Soc Trans. 1991 Apr;19(2):368-73.
[59] Boppart MD, Hirshman MF, Sakamoto K, Fielding RA, Goodyear LJ. Static stretch increases c-Jun NH2-terminal kinase activity and p38 phosphorylation in rat skeletal muscle. Am J Physiol Cell Physiol. 2001 Feb;280(2):C352-8.
[60] Koj A. Initiation of acute phase response and synthesis of cytokines. Biochim Biophys Acta. 1996 Nov 15;1317(2):84-94.
[61] Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995 Mar 31;270(13):7420-6.
[62] Force T, Bonventre JV. Growth factors and mitogen-activated protein kinases. Hypertension. 1998 Jan;31(1 Pt 2):152-61.
[63] Kyriakis JM, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays. 1996 Jul;18(7):567-77.
[64] Pillinger MH, Capodici C, Rosenthal P, Kheterpal N, Hanft S, Philips MR, Weissmann G. Modes of action of aspirin-like drugs: salicylates inhibit erk activation and integrin-dependent neutrophil adhesion. Proc Natl Acad Sci U S A. 1998 Nov 24;95(24):14540-5.
[65] Frost RA, Nystrom GJ, Lang CH. Tumor necrosis factor-alpha decreases insulin-like growth factor-I messenger ribonucleic acid expression in C2C12 myoblasts via a Jun N-terminal kinase pathway. Endocrinology. 2003 May;144(5):1770-9.
Thanks,